 Genome Technologies Laboratory Home
Genome Technologies Laboratory Home
This is the home of the Genome Technologies Laboratory, a shared University of Wyoming lab that supports analyses of genomes. The lab is located in the Berry Center in Room 320, is staffed by @Gregg Randolph and advised by Nic Blouin. Please use the sidebar to the left to navigate the structure of this website.
Services:
DNA library prep for Illumina sequencing
16S (bacterial barcoding), ITS(fungus), COI(animals), 18S(designed for arbuscular mycorhyzal fungus), TRNL (herbivore diet barcoding), and CO1 (designed for insectivorous diet studies) amplicon library preps with high levels of multiplexing
ddRAD GBS for UWyo Associated clients only
Other libraries preps for Illumina sequencing
DNA extraction using Qiagen kits
Consulting
Oxford Nanopore MinIon prep and sequencing
We regularly prepare DNA sequencing libraries for Illumina instruments. We either sequence on campus on a NextSeq 2000 or send samples out to a commercial provider we have negotiated good pricing on NovaSeq X runs. For pilot or other small scale projects, we can sequence Illumina libraries on an iSeq instrument. We have robotic liquid handling and other equipment for high throughput DNA extractions using Qiagen kits and DNA sequencing library preparation.
Information regarding the GTL
https://microcollaborative.atlassian.net/wiki/spaces/MICLAB/pages/1732182029
Standard operating procedures
Equipment in the lab includes
ABI 7500 Real Time PCR System (quantitative PCR)
Agilent 2100 Bioanalyzer (Fragment Analyzer)
Bio Rad C1000 Touch Thermocyclers
Biotek Synergy HTX (multimode plate reader)
Hamilton Microlab Nimbus (Automated Liquid Handling - Library Prep)
Illumina iSeq 100 (small project DNA sequencing)
Integra Biosciences Assist Plus (Automated Liquid Handling - Extraction)
Nanodrop ND-1000 (Spectrophotometer-protein and nucleic acid quantification)
Oxford Nanopore MinIon (A third generation sequencer for longer reads)
Savant Speedvac DNA 130 (Vacuum concentrator)
Pages for use by GTL staff and micro project users
Lab rules - all users of the GTL space and equipment should read these rules prior to beginning work.
GTL projects, both ongoing and completed (under construction)
https://microcollaborative.atlassian.net/wiki/spaces/GTLI/pages/546144480
A common question in sequencing relates to the diminishing returns of sequencing the same material to greater and greater depth and at what point our uncertainty about the frequency of a component has diminished sufficiently (an allele at a locus, or a taxonomic unit in an environmental sample), given the trade-off between sequencing more at this locus rather than sequencing more samples or genomic regions.
We can simplify this problem to wanting to know the frequency of an allele or taxon, and the frequency of everything else (the complement). This binomial categorization is a simplification of the multinomial and has everything we need (there is no gain in considering the frequencies of other categories for this illustration or in general for knowing the frequency of a component of a composition; the binomial model is entirely representative of those multinomial models).
The greatest uncertainty exists when the category of interest is at frequency 0.5, so I present that as an upper bound in the plots below. I also illustrate the uncertainty for a frequency of 0.05, and the diminishing returns on sequencing at greater depth for that sample. For lower frequency components the uncertainty would be even smaller.
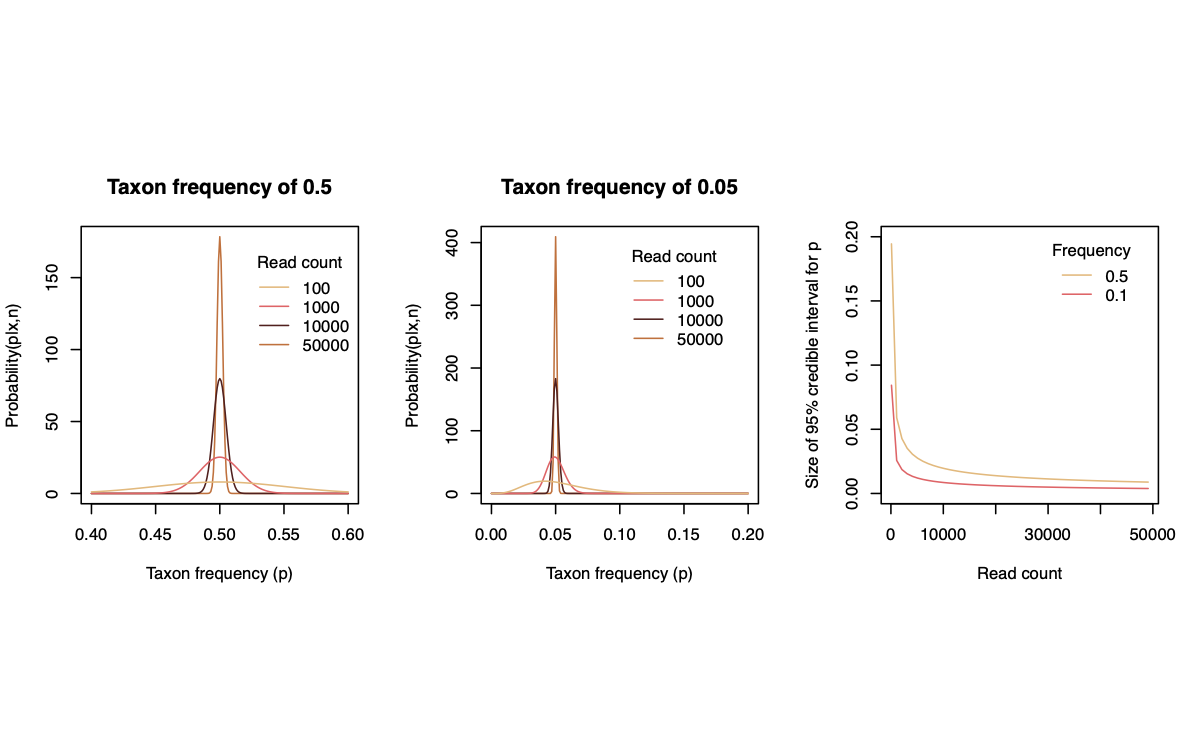
Here is the code should you want to play with this.
Joshua HarrisonGregg Randolph and Alex Buerkle have published an open-access paper in mSystems that reviews technologies for amplicon sequencing and presents context and rationales for the standard methods that we use for 16S and ITS preps in the https://microcollaborative.atlassian.net/wiki/spaces/MICLAB.
Harrison JG, Randolph GD, Buerkle CA. 2021. https://doi.org/10.1128/mSystems.00294-21 mSystems 6:e00294-21.
New approaches to characterizing microbiomes via high-throughput sequencing provide impressive gains in efficiency and cost reduction compared to approaches that were standard just a few years ago. However, the speed of method development has been such that staying abreast of the latest technological advances is challenging. Moreover, shifting laboratory protocols to include new methods can be expensive and time consuming. To facilitate adoption of new techniques, we provide a guide and review of recent advances that are relevant for single-locus sequence-based study of microbiomes—from extraction to library preparation—including a primer regarding the use of liquid-handling automation in small-scale academic settings. Additionally, we describe several amendments to published techniques to improve throughput, track contamination, and reduce cost. Notably, we suggest adding synthetic DNA molecules to each sample during nucleic acid extraction, thus providing a method of documenting incidences of cross-contamination. We also describe a dual-indexing scheme for Illumina sequencers that allows multiplexing of many thousands of samples with minimal PhiX input. Collectively, the techniques that we describe demonstrate that laboratory technology need not impose strict limitations on the scale of molecular microbial ecology studies.
IMPORTANCE New methods to characterize microbiomes reduce technology-imposed limitations to study design, but many new approaches have not been widely adopted. Here, we present techniques to increase throughput and reduce contamination alongside a thorough review of current best practices.
I thought that Noah Fierer was going to write about the hidden cost of bioinformatics and other analysis in his blog post “The ‘hidden’ costs of microbial community analyses“, but instead he points out that most of the cost of the laboratory analysis is not necessarily in the sequencing. This will be even more true for our project, where we plan to do an order of magnitude higher levels of multiplexing within a lane of sequencing. Someone else will need to evaluate the cost of the downstream, computational analysis of the sequences.